
Warning
This documentation is for CPAT v2.0.0 or older versions. For documentation of CPAT v3.0.0 and future versions, please go to https://cpat.readthedocs.io/en/latest/
Release history¶
CPAT v2.0.0
This version and all future versions will run in Python3. Installation use “pip3 install CPAT”
CPAT v1.2.4
Supports installation using pip
Fix inconsistence in LISCENSE.
CPAT v1.2.3
v1.2.3 is almost identical to v1.2.2, with only one change: In CPAT v1.2.2 (line 28), “import pysam” was unintentionally commented out. This error is fixed in v1.2.3.
CPAT v1.2.2
Fixed bugs in longest_orf() function. In CPAT (v1.2.2), if a mRNA sequence contains two ORFs with the same length, it reports the one that is upstream (i.e. closer to transcript start site). While previous version reported the one that detected first. For example:
This sequence contains two very simple open reading frames quoted by “
5’-A “ATGTAG” AA “ATGCTG”-3’
Old version will return “ATGCTG” simply because it uses the first reading frame, while current version will return “ATGTAG” because it’s closer to transcription start site.
Thanks Kristoffer Vitting-Seerup from University of Copenhagen for reporting this bug.
CPAT v1.2.1
CPAT v1.2
Conservation score was obsolete. Because it depends on the alignment, relatively slow in calculation and more importantly very little power is gained by using this feature. We use hexamer usage bias as the 4th feature:
ORF (Open Reading Frame) size
ORF coverage (ratio of ORF size to transcript size)
Fickett TESTCODE statistic
Hexamer usage bias
CPAT v1.1:
This is the only version using conservation score (phastCon) as one prediction feature. 4 features used for prediction:
ORF (Open Reading Frame) size
ORF coverage (ratio of ORF size to transcript size)
Fickett TESTCODE statistic
PhastCon conservation score
Introduction¶
Using RNA-seq, tens of thousands of novel transcripts and isoforms have been identified (Djebali, et al Nature, 2012 , Carbili et al, Gene & Development, 2011) The discovery of these hidden transcriptome rejuvenate the need of distinguishing coding and noncoding RNA. However, Most previous coding potential prediction methods heavily rely on alignment, either pairwise alignment to search for protein evidence or multiple alignments to calculate phylogenetic conservation score (such as CPC , PhyloCSF and RNACode ). This is because most previously identified transcripts including protein coding RNA and short, housekeeping/regulatory RNAs such as snRNAs, snoRNA and tRNA are highly conserved. While still very useful, these approaches have several limitations:
Most lncRNAs are less conserved and tend to be lineage specific which greatly limit the discrimination power of alignment-based methods. For example, of 550 lncRNAs detected from zebrafish, only 29 of them had detectable sequence similarity with putative mammalian orthologs (Ulitsky et al, Cell, 2011).
A significant fraction of protein coding genes may have an alternatively processed isoform or one transcribed from an alternative promoter, these part of ncRNA cannot be correctly classified through homologous search because they would have significant match to protein coding genes.
Alignment based method is extremely slow. For example, CPC takes 6050 CPU minutes (> 4 days) to evaluate 14,000 lncRNA transcripts.
Reliability depends on alignment quality. Most multi-alignment tools use heuristic search and do not guarantee to give optimal alignments.
CPAT overcomes the above issues by using logistic regression model based on 4 pure sequence-based, linguistic features
ORF size
ORF coverage
Linguistic features based method does not require other genomes or protein databases to perform alignment and is more robust. Because it is alignment free, it runs much faster and also easier to use. For example, CPAT only took several minutes to evaluate the above 14,000 lncRNAs. More importantly, compared with alignment-based approaches, CPAT achieves better sensitivity and specificity (0.966 tested on human gene annotation).
Run CPAT online¶
https://wlcb.oit.uci.edu/cpat is hosted by Dr Wei Li’s lab @ University of California Irvine.
Step1: Upload data to CPAT server. There are 3 different ways to uploada
Upload BED or FASTA format files from local disk. Files can be regular or compressed (*.gz, *.Z. *.z, *.bz, *.bz2, *.bzip2).
For small dataset, user can copy and paste data (in BED or FASTA format) directly to the text area.
For extremely larger dataset, user can save data in web server (http, https or ftp) first, then paste the data url to text area.
Step2: Select Select Species assembly
Step3: Click Submit button
NOTE
This web server only supports Human (hg19), Mouse (mm9 and mm10), Fly (dm3) and Zebrafish (Zv9).
When input file is BED format, the reference genome is required and the assembly version is important. For example, user cannot upload hg18 based BED file to this server, as we only support hg19.
When the input file is FASTA sequence file, the reference genome is NOT required, because all features can be calculated from the FASTA sequence directly.
FASTA sequence file is supported but not recommended, because if the file size is too large, user may encounter ‘time out’ error. If you only have FASTA file, compressed it using gzip or bunzip2 before uploading. Or, you can save it to a file server and provide the url to CPAT.
Install CPAT to local computer¶
Installation for CPAT (version 1.2.4)¶
Prerequisite: python2.7; numpy; R:
# install CPAT hosted on PyPI
$ pip2 install CPAT
#or you can download CPAT-VERSION.tar.gz first and then run:
$ pip2 install CPAT-VERSION.tar.gz
Installation for CPAT (version 2.0.0 or later version)¶
Prerequisite: python3.5 or later version; numpy; R:
# install CPAT hosted on PyPI
$ pip3 install CPAT
#or you can download CPAT-VERSION.tar.gz first and then run:
$ pip3 install CPAT-VERSION.tar.gz
NOTE:
User need to download prebuilt logit model and hexamer table for human, mouse, zebrafish and fly. For other species, we provide scripts to build these models (see below).
If you had “error: invalid command ‘egg_info’” when running ‘pip install CPAT-VERSION.tar.gz’. Try to upgrade setuptools:
$ pip install –upgrade setuptools
Command line usage¶
Input file¶
BED format file (regular text or compressed). BED file should be in standard 12-column format.
FASTA format file (regular text or compressed)
a URL pointing to data that are saved remotely (data could be either BED or FASTA, either regular text or compressed file). http://, https:// and ftp:// are supported.
cpat.py¶
User need to provide a gene file (‘-g’), a logit model file (‘-d’), a hexamer frequency table file (‘-x’) and specify the output file name(‘-o’). Gene file could be either in BED or FASTA format. If in BED format, user also needs to specify reference genome sequence (‘-r’); if gene file is in fasta format, each mRNA sequence must be in 5’->3’ direction.
- Options:
- --version
show program’s version number and exit
- -h, --help
show this help message and exit
- -g GENE_FILE, --gene=GENE_FILE
Transcripts either in BED format or mRNA sequences in FASTA format: If this is BED format file, ‘-r’ must be specified; if this is mRNA sequence file in FASTA format, ignore the ‘-r’ option. The input BED or FASTA file could be regular text file or compressed file (*.gz, *.bz2) or accessible url.
- -o OUT_FILE, --outfile=OUT_FILE
output file. Tab separated text file: geneID <tab> mRNA size <tab> ORF size <tab> Fickett Score <tab> Hexamer Score<tab>Coding Probability.
- -x HEXAMER_DAT, --hex=HEXAMER_DAT
Prebuilt hexamer frequency table (Human, Mouse, Fly, Zebrafish). Run ‘make_hexamer_tab.py’ to make this table out of your own training dataset.
- -d LOGIT_MODEL, --logitModel=LOGIT_MODEL
Prebuilt training model (Human, Mouse, Fly, Zebrafish). Run ‘make_logitModel.py’ to build logit model out of your own training datset
- -r REF_GENOME, --ref=REF_GENOME
Reference genome sequences in FASTA format. Ignore this option if mRNA sequences file was provided to ‘-g’. Reference genome file will be indexed automatically (produce *.fai file along with the original *.fa file within the same directory) if hasn’t been done.
- -s START_CODONS, --start=START_CODONS
Start codon (DNA sequence, so use ‘T’ instead of ‘U’) used to define open reading frame (ORF). default=ATG
- -t STOP_CODONS, --stop=STOP_CODONS
Stop codon (DNA sequence, so use ‘T’ instead of ‘U’) used to define open reading frame (ORF). Multiple stop codons should be separated by ‘,’. default=TAG,TAA,TGA
Examples 1:
$ cd CPAT-1.2.2/test
# use BED file as input. '-r' is required
$ cpat.py -r /database/hg19.fa -g Human_test_coding_mRNA_hg19.bed -d ../dat/Human_logitModel.RData -x ../dat/Human_Hexamer.tsv -o output1
# use FASTA file as input. '-r' is not required
$ cpat.py -g Human_test_coding_mRNA.fa -d ../dat/Human_logitModel.RData -x ../dat/Human_Hexamer.tsv -o output2
$ head output1
mRNA_size ORF_size Fickett_score Hexamer_score coding_prob
NM_198317 2564 1929 1.1902 0.580124008851 0.999999999400781
NM_001014980 1043 909 1.1934 0.495124501575 0.999917485272474
NM_004421 2924 2013 1.0965 0.584852243126 0.999999999679495
NM_032348 2273 1329 1.2568 0.596113918225 0.999999649979388
NM_022834 4659 1338 1.1722 0.5589769999 0.999999395067735
NM_199121 4264 702 1.1722 0.560116277167 0.999276921874284
NM_001170535 2502 1761 1.2286 0.638700023819 0.999999997598248
NM_018188 2646 1905 1.2286 0.611789944749 0.999999999430874
NM_001170536 2332 1524 1.2712 0.661478359365 0.999999974636409
$ head output2
mRNA_size ORF_size Fickett_score Hexamer_score coding_prob
HG19_CT_USERTRACK_3545_NM_173083 3345 1677 0.7983 0.0216287267657 0.999998800005244
HG19_CT_USERTRACK_3545_NM_002744 2326 1779 1.2302 0.495437684259 0.999999995386405
HG19_CT_USERTRACK_3545_NM_001033581 2114 1230 1.3191 0.537935154638 0.999998793893276
HG19_CT_USERTRACK_3545_NM_001033582 2011 1230 1.3191 0.537935154638 0.999998800685703
HG19_CT_USERTRACK_3545_NM_032409 2660 1746 1.0164 0.361862240133 0.99999996805242
HG19_CT_USERTRACK_3545_NM_032236 4464 3108 0.9865 0.156660830868 1
HG19_CT_USERTRACK_3545_NM_130440 7706 5697 1.2799 0.579103525916 1
HG19_CT_USERTRACK_3545_NM_002840 7733 5724 1.2799 0.579964454487 1
HG19_CT_USERTRACK_3545_NM_007051 2583 1953 0.9702 0.184701969294 0.999999989213307
make_hexamer_tab.py¶
make_hexamer_tab.py calculates the in frame hexamer (6mer) frequency from CDS sequence in fasta format. The CDS is mRNA sequence that removes UTR. This table is required by CPAT to calculate the hexamer usage score. Users can download prebuilt hexamer tables (Human, Mouse, Fly, Zebrafish) from here
- Options:
- --version
show program’s version number and exit
- -h, --help
show this help message and exit
- -c CODING_FILE, --cod=CODING_FILE
Coding sequence (must be CDS without UTR, i.e. from start coden to stop coden) in fasta format. User can get CDS sequence of a bed file using UCSC table browser
- -n NONCODING_FILE, --noncod=NONCODING_FILE
Noncoding sequences in fasta format
Example:
$ cd CPAT-1.2.2/test
$ make_hexamer_tab.py -c Human_coding_transcripts_CDS.fa -n Human_noncoding_transcripts_RNA.fa >Human_Hexamer.tsv
$ head Human_Hexamer.tsv
hexamer coding noncoding
GAACGT 0.000114999540425 6.20287252729e-05
CTTCTT 0.000280298143192 0.000464526231488
CACCCT 0.000254883880114 0.000337895737524
GAACGG 0.000178535198119 5.8077265737e-05
GAACGC 0.000136389878516 6.03746259323e-05
GAACGA 0.00015830968042 5.87205265917e-05
CACCCA 0.000258696019576 0.000448628498937
CTTCTA 0.000147508618612 0.000280645521457
CACCCC 0.000328479350276 0.000342582352322
...
make_logitModel.py¶
Build logistic regression model (“prefix.logit.RData”) required by CPAT. This program will output 3 files:
prefix.feature.xls: A table contains features calculated from training datasets (coding and noncoding gene lists).
prefix.logit.RData: logit model required by CPAT (if R was installed).
prefix.make_logitModel.r: R script to build the above logit model.
Note: Users can download prebuilt logit models (Human, Mouse, Fly, Zebrafish) from here
- Options:
- --version
show program’s version number and exit
- -h, --help
show this help message and exit
- -c CODING_FILE, --cgene=CODING_FILE
Protein coding transcripts (used to build logit model) either in BED format or mRNA sequences in FASTA format: If this is BED format file, ‘-r’ must be specified; if this is mRNA sequence file in FASTA format, ignore the ‘-r’ option. The input BED or FASTA file could be regular text file or compressed file (*.gz, *.bz2) or accessible url. NOTE: transcript ID should be unique.
- -n NONCODING_FILE, --ngene=NONCODING_FILE
Non protein coding transcripts (used to build logit model) either in BED format or mRNA sequences in FASTA format: If this is BED format file, ‘-r’ must be specified; if this is mRNA sequence file in FASTA format, ignore the ‘-r’ option. The input BED or FASTA file could be regular text file or compressed file (*.gz, *.bz2) or accessible url. NOTE: transcript ID should be unique.
- -o OUT_FILE, --outfile=OUT_FILE
output prefix.
- -x HEXAMER_DAT, --hex=HEXAMER_DAT
Prebuilt hexamer frequency table (Human, Mouse, Fly, Zebrafish). Run ‘make_hexamer_tab.py’ to generate this table.
- -r REF_GENOME, --ref=REF_GENOME
Reference genome sequences in FASTA format. Ignore this option if mRNA sequences file was provided to ‘-g’. Reference genome file will be indexed automatically (produce *.fai file along with the original *.fa file within the same directory) if hasn’t been done.
- -s START_CODONS, --start=START_CODONS
Start codon (DNA sequence, so use ‘T’ instead of ‘U’) used to define open reading frame (ORF). default=ATG
- -t STOP_CODONS, --stop=STOP_CODONS
Stop codon (DNA sequence, so use ‘T’ instead of ‘U’) used to define open reading frame (ORF). Multiple stop codons should be separated by ‘,’. default=TAG,TAA,TGA
Example:
$ cd CPAT-1.2.2/test
$ make_logitModel.py -x Human_Hexamer.tsv -c Human_coding_transcripts_mRNA.fa -n Human_noncoding_transcripts_RNA.fa -o Human
Process protein coding transcripts: Human_coding_transcripts_mRNA.fa
Input gene file is in FASTA format
Process non coding transcripts: Human_noncoding_transcripts_RNA.fa
Input gene file is in FASTA format
build logi model ...
Warning message:
glm.fit: fitted probabilities numerically 0 or 1 occurred
#or use BED file as input
$ make_logitModel.py -x Human_Hexamer.tsv -c Human_coding_transcripts_hg19.bed -n Human_noncoding_transcripts_hg19.bed -r /database/hg19.fa -o Human
How to choose cutoff¶
Optimum cutoff were determined from TG-ROC.
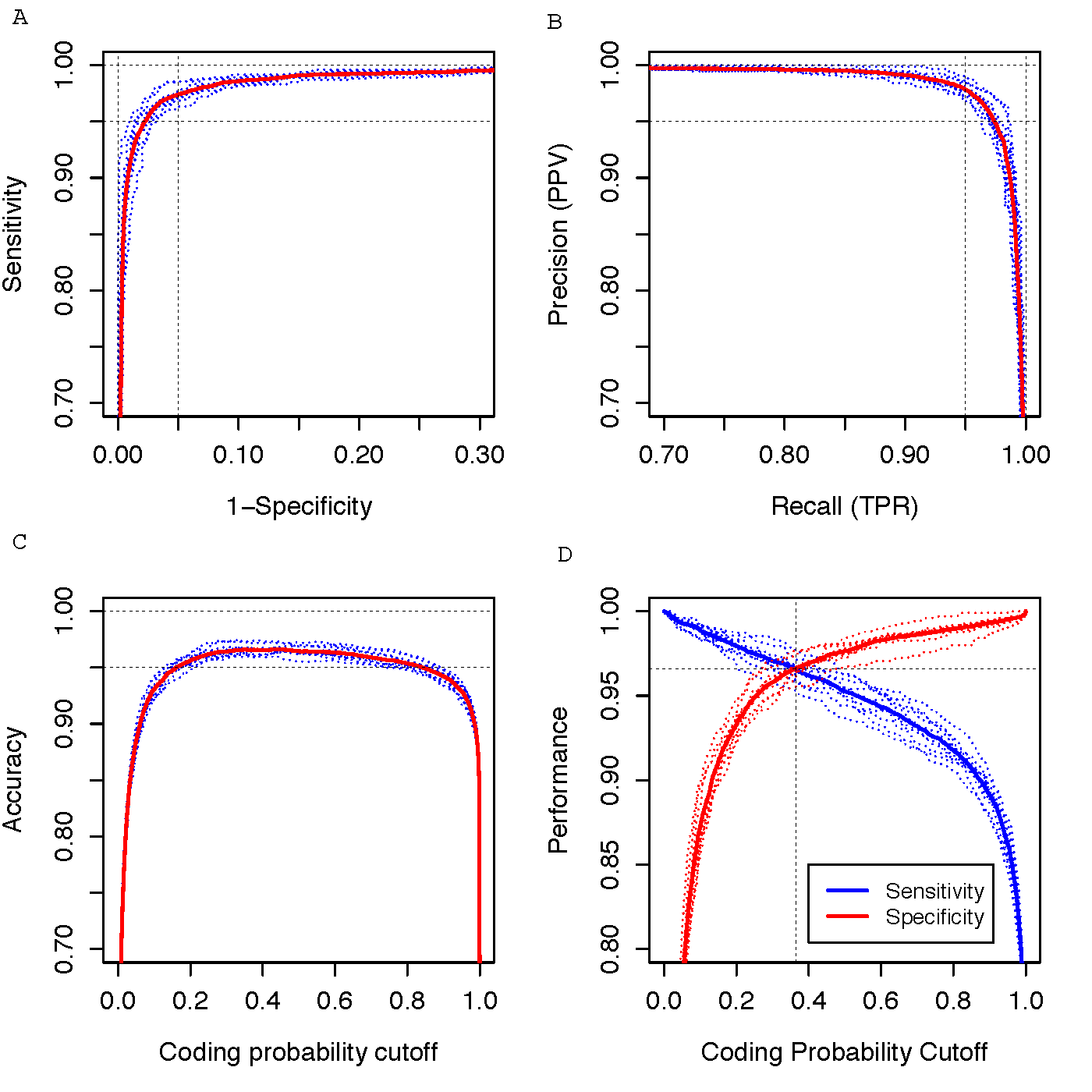
Human coding probability (CP) cutoff: 0.364 (CP >=0.364 indicates coding sequence, CP < 0.364 indicates noncoding sequence) (see performance figure D)
Mouse coding probability (CP) cutoff: 0.44
Fly coding probability (CP) cutoff: 0.39
Zebrafish coding probability (CP) cutoff: 0.38
Here we provide the R code and the data that we used to generate Figure 3 in our paper. Note the ROCR library is required to run our R code.
1) Download R code and data from here
2) Put the R code and the data table in the same folder
$ ls
10Fold_CrossValidation.r Human_train.dat
3) Run the R code from command line or console. The R code will perform 10-fold cross validation and generate Figure_3.
$ Rscript 10Fold_CrossValidation.r # install ROCR before running this code
Loading required package: gplots
Attaching package: ‘gplots’
The following object is masked from ‘package:stats’:
lowess
Loading required package: methods
Warning message:
package ‘gplots’ was built under R version 3.1.2
[1] "ID" "mRNA" "ORF" "Fickett" "Hexamer" "Label"
Warning message:
glm.fit: fitted probabilities numerically 0 or 1 occurred
Warning message:
glm.fit: fitted probabilities numerically 0 or 1 occurred
Warning message:
glm.fit: fitted probabilities numerically 0 or 1 occurred
Warning message:
glm.fit: fitted probabilities numerically 0 or 1 occurred
Warning message:
glm.fit: fitted probabilities numerically 0 or 1 occurred
Warning message:
glm.fit: fitted probabilities numerically 0 or 1 occurred
Warning message:
glm.fit: fitted probabilities numerically 0 or 1 occurred
Warning message:
glm.fit: fitted probabilities numerically 0 or 1 occurred
Warning message:
glm.fit: fitted probabilities numerically 0 or 1 occurred
Warning message:
glm.fit: fitted probabilities numerically 0 or 1 occurred
null device
1
How to prepare training dataset¶
We prebuild hexamer tables and logit models for human, mouse, fly and zebrafish. If you want to run CPAT for other species, you need to prepare “coding sequences” and “noncoding sequences” as training data. These two files are required when you run make_hexamer_tab.py and make_logitModel.py.
Coding sequences: the whole CDS part of the mRNA. In other words, each CDS sequences should start with start codon and end with stop coden. As you expected, the length of each CDS should be an integer multiple of 3. You should reverse-compliment the CDS sequence if the mRNA sequence is extracted from the genome and the gene is located on ‘-‘ strand.
Noncoding sequences: It’s better to use those annotated (known) noncoding genes, rather than the “noncoding parts” of protein coding genes such as 3’UTR and 5’UTR.
It’s better to have balanced training dataset (i.e. the number of coding sequences is roughly equal to the number of noncoding sequences).
If the genome of the species you are working on is NOT well annotated and does not have enough “coding” and “noncoding” genes to build the training data, you could build your model using data from other species that is evolutionary close to the species you are working on.
Evaluating Performance¶
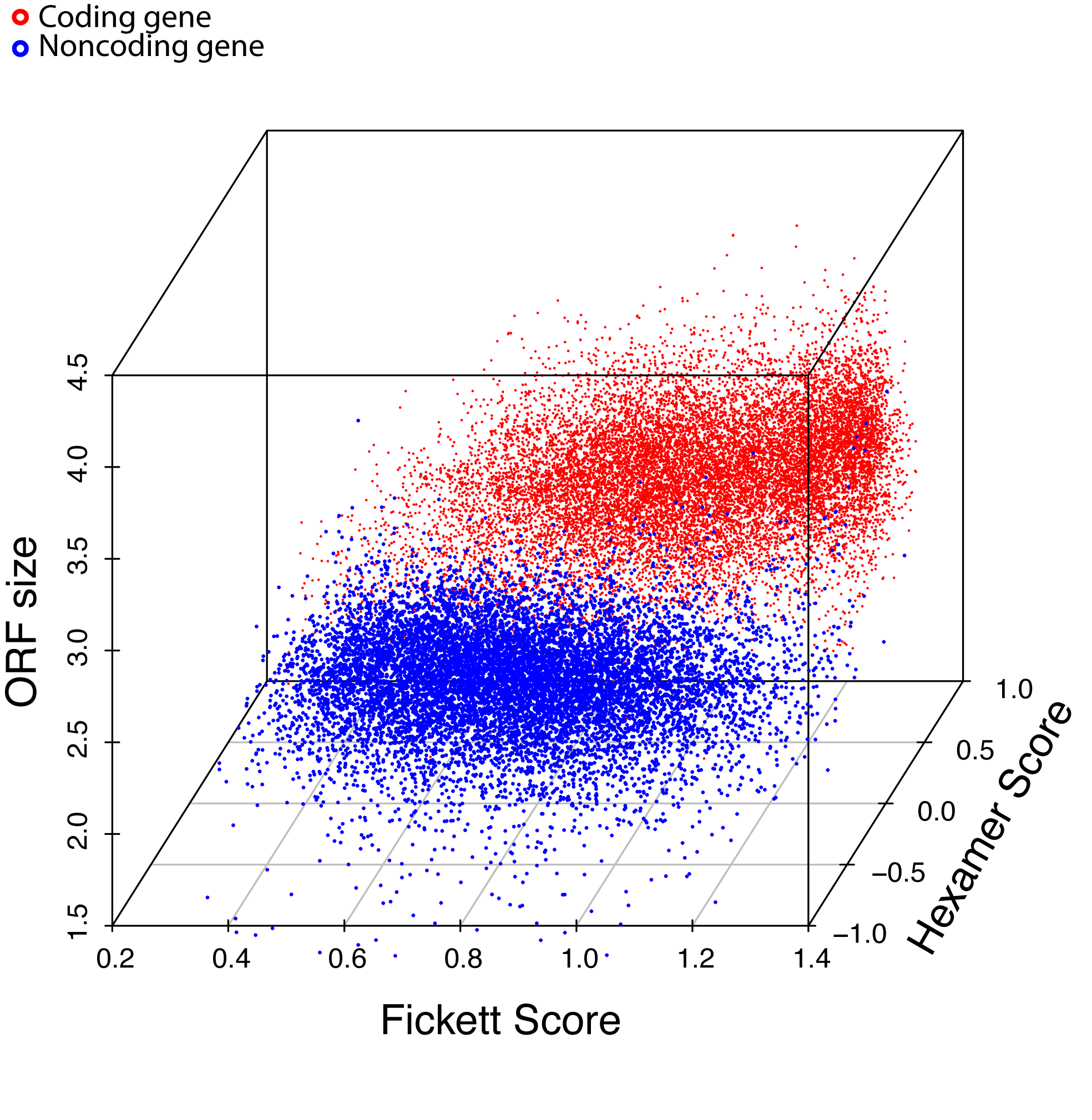
Combinatorial effects of 3 major features. 10,000 coding genes (red dots) and 10,000 noncoding genes (blue dots) are clearly separated into two clusters. (below figure)
Performance evaluation using 10-fold cross validation (10,000 coding genes and 10,000 noncoding genes). Blue dotted curves represent the 10-fold cross validations, red solid curve represents the averaged curve between 10 runs of validations. (A) ROC curve. (B) PR (precision-recall) curve. (C) Accuracy vs cutoff value. (D) Two graphic ROC curve to determine the optimum cutoff value.
Comparison¶
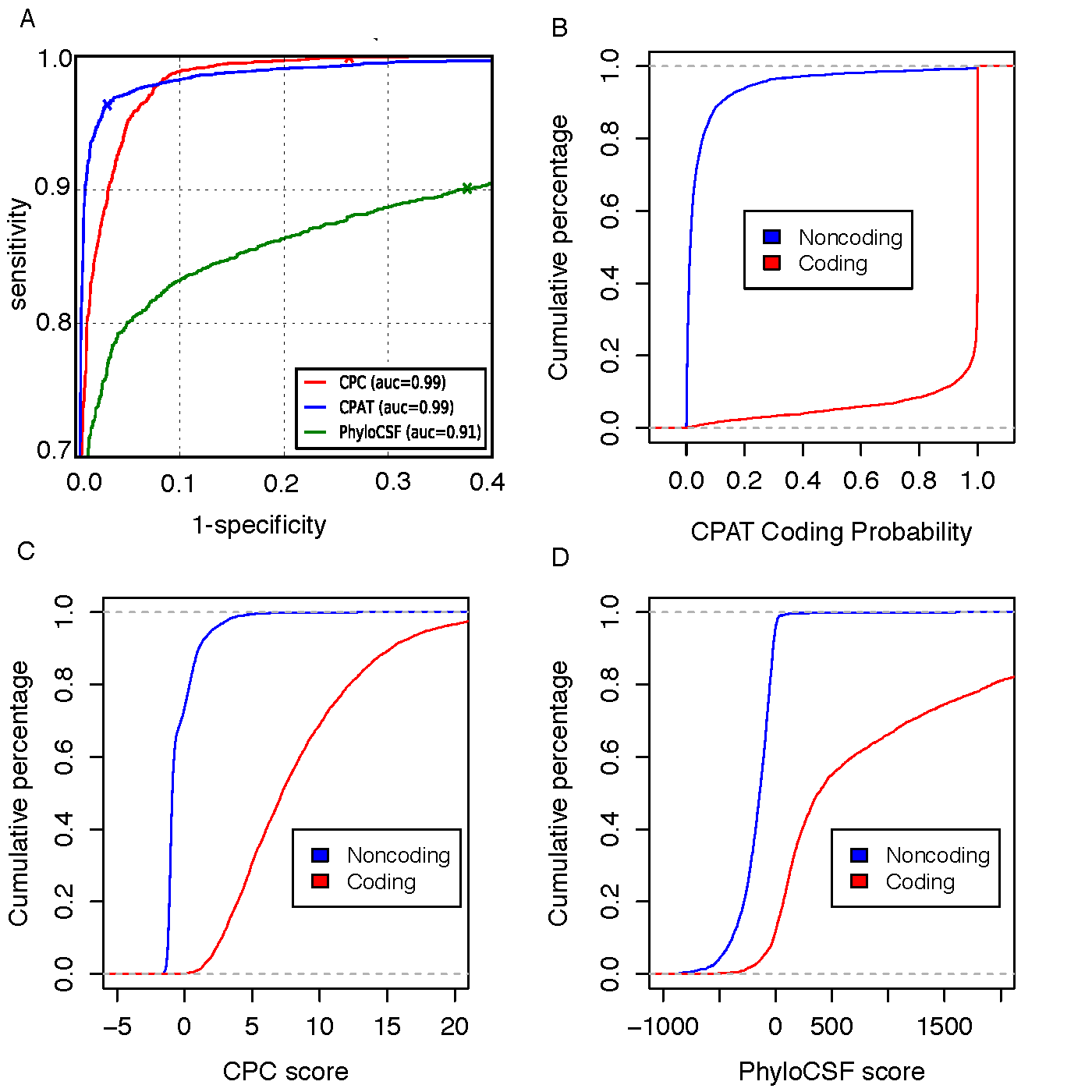
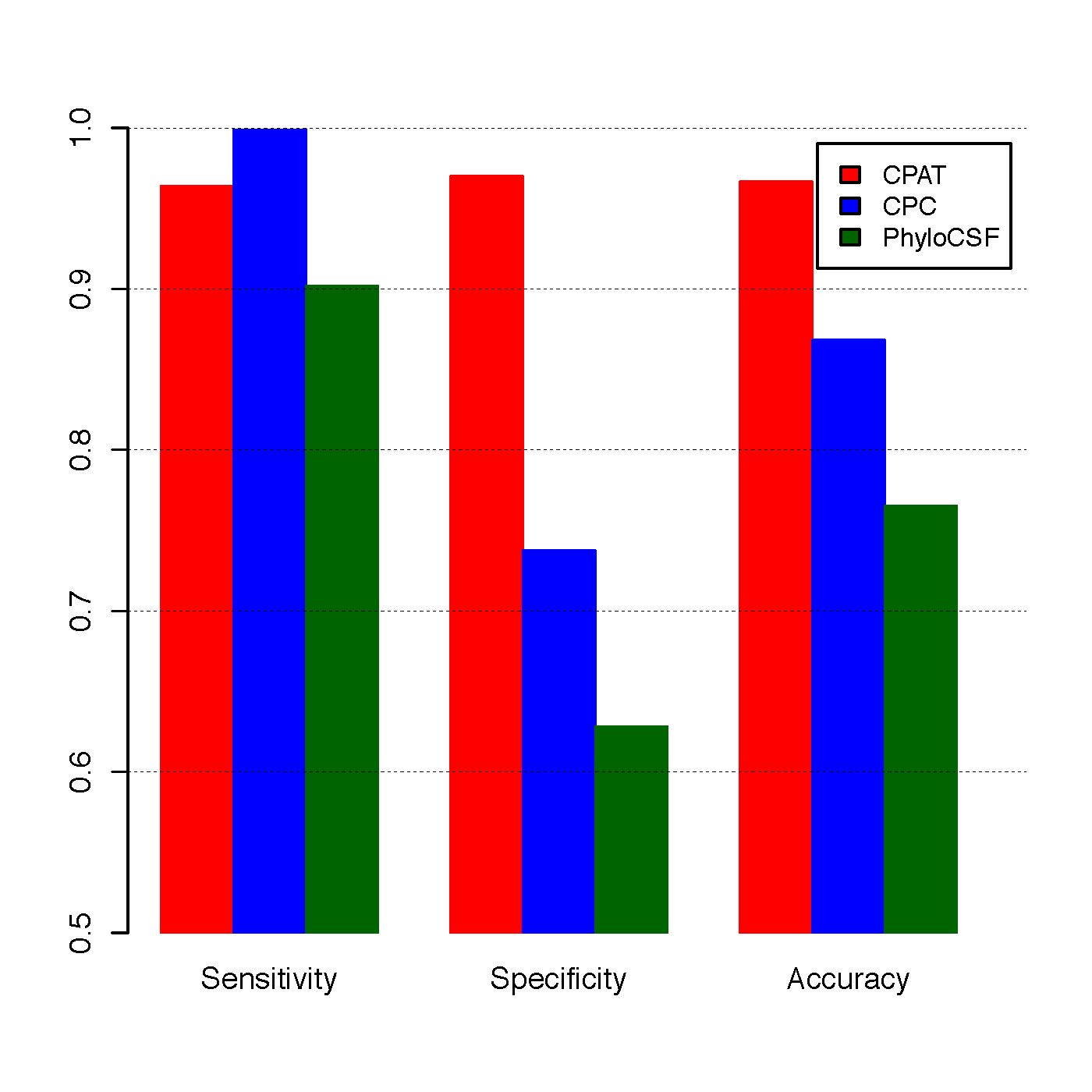
To compare CPAT with CPC and PhyloCSF, we build an independent testing dataset that composed of 4,000 high quality protein coding genes from Refseq annotation and 4,000 lincRNAs from Human lincRNA catalog (Cabili et al., 2011). All 8000 genes were not included in the training dataset of CPAT.
LICENSE¶
CPAT is distributed under GNU General Public License
This program is free software; you can redistribute it and/or modify it under the terms of the GNU General Public License as published by the Free Software Foundation; either version 2 of the License, or (at your option) any later version. This program is distributed in the hope that it will be useful, but WITHOUT ANY WARRANTY; without even the implied warranty of MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. See the GNU General Public License for more details. You should have received a copy of the GNU General Public License along with this program; if not, write to the Free Software Foundation, Inc., 51 Franklin Street, Fifth Floor, Boston, MA 02110-1301 USA
Reference¶
Wang, L., Park, H. J., Dasari, S., Wang, S., Kocher, J.-P., & Li, W. (2013). CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Research, 41(6), e74. doi:10.1093/nar/gkt006
Contact¶
Liguo Wang: wang.liguo AT mayo.edu
Wei Li: wei.li AT uci.edu